
引言
本系列讲解 空间转录组学 (Spatial Transcriptomics) 相关基础知识与数据分析教程,持续更新,欢迎关注,转发,文末有交流群!
背景
基于测序的空间转录组学(ST)平台通过使用下一代测序(NGS)技术,结合空间条形码,在组织的不同空间位置上对基因表达进行定量分析。在平台制造和样本制备过程中,空间位置信息被编码,并与测序过程中检测到的转录本相关联。这种关联体现在NGS平台测序生成的序列读取结构中。
为了进行空间数据分析,需要将原始测序数据经过一系列预处理步骤,转化为有用的数据格式,通常是计数矩阵。通过计数矩阵,我们可以分析目标组织中的基因表达情况。这些预处理步骤因平台而异,但基本流程是从一系列“读取序列”开始,最终生成适用于下游分析工具(如Squidpy、Seurat或基于SpatialExperiment对象的Bioconductor工作流程)的空间数据格式。
序列和测序
在转录组学中,“读取序列”是指从RNA分子逆转录而来的cDNA片段的核苷酸序列。这些转录本的丰度反映了基因表达水平,而这正是转录组学分析的核心目标。空间转录组学的优势在于能够将读取序列与RNA分子的起源位置相关联,从而揭示基因表达的空间分布。生成读取序列的过程通常包括以下几个步骤:
- RNA提取
- 逆转录
- cDNA片段化
- 接头连接
- PCR扩增
由于PCR扩增步骤的存在以及RNA提取过程中的不完美性,读取序列的丰度只能作为基因表达的相对指标,而不能作为绝对值。因此,在进行差异表达分析等下游分析之前,需要对数据进行归一化处理。在归一化之前,读取序列需要经过一系列预处理步骤,构建计数矩阵或其他等效数据结构,以便用于后续分析。
序列结构
在大多数基于测序的空间技术中,读取序列通常以“配对末端”的形式记录,即DNA片段的两端分别被测序,并分别存储在不同的文件中,通常是.fastq格式的文件。其中一个文件(通常是读取1)包含条形码序列,而根据是否事先对读取序列进行了修剪,它还可能包含连接序列或其他结构序列。另一个文件则包含我们需要与参考基因组或转录组(或探针集)进行比对以确定表达基因的转录本(或探针)序列。
以下是BGI STOmics Stereo-seq用户手册中提供的一个读取序列结构示例:
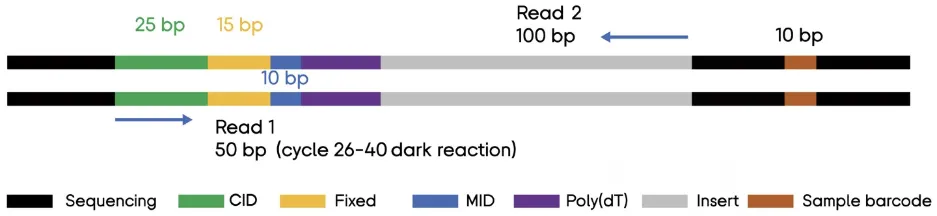
在这里,我们可以看到读取1是从序列左端开始的前50bp,而读取2是从序列右端开始的最后100bp。
读取1中包含了25bp的坐标ID(CID)、一个15bp的固定连接序列,以及一个10bp的分子ID(MID)。
读取2仅包含一个100bp长的转录本片段。
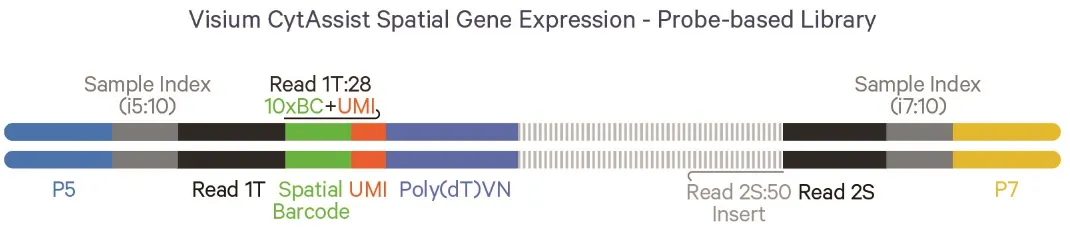
另一个例子来自10X Visium CytAssist试剂盒,用于展示基于探针的文库的结构:
在.fastq文件中,每个读取序列都包含一个对应的序列标题和质量评分。这里提供了一个示例(同样来自BGI STOmics Stereo-seq用户手册),用于说明:
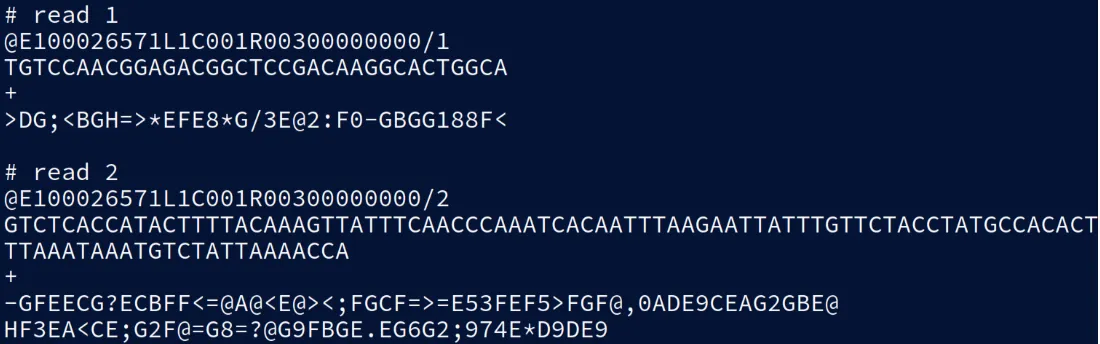
两条读取序列的第一行是“标题”或“名称”,用于唯一标识每条读取序列,并可能包含一些额外信息,例如读取序列来自测序仪的哪一条泳道。此外,标题部分还可以插入工具生成的附加元数据,以“注释”的形式呈现。第二行是测序转录本的碱基序列,如前文所述。第三行是一个间隔行,通常只包含一个“+”字符,尽管有时会在这里重复标题中的读取序列标识符和注释。第四行是序列中每个碱基的读取质量评分。质量评分的衡量标准会因测序仪的版本以及是否使用Q4或Q40文件而有所不同。Q分数是基于p值的对数形式,用于衡量对碱基判定的置信度。p值的确切计算方法以及读取序列被判定为不可靠的阈值因平台而异,因此如果这些统计信息对你的分析很重要,建议仔细检查你所使用的工具。





