
简介
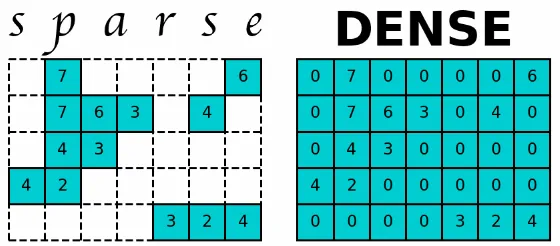
为了更好地处理、存储以及可视化高分辨率的 Hi-C 数据,开发出了高效的数据格式。其中,只保存非零值的稀疏矩阵格式得到了广泛应用,成为了行业标准。此外,“zoomable”矩阵风格也受到了青睐,它可以在一个文件里存储不同分辨率的数据,方便进行交互式可视化。本文主要介绍了像 .hic 和 .mcool 这样的最新矩阵文件格式,以及 SAM/BAM 和可随机访问的接触列表等其他中间格式。
引言
原位 Hi-C 技术的出现,使得 Hi-C 图谱的分辨率从兆碱基对提升到了千碱基对级别。随着数据规模的大幅增加,人们迫切需要开发出更高效的格式来应对。
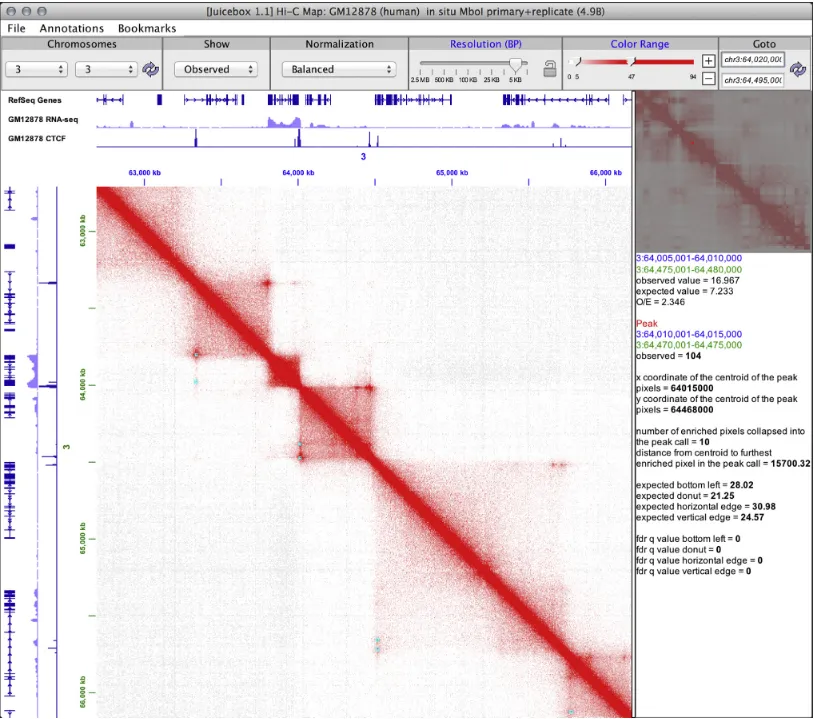
举例来说,一个千碱基对分辨率的全基因组相互作用矩阵,会包含数百亿个元素,而且需要处理多达数十亿对基因组位置。 .hic 矩阵格式和其他中间格式,最早是在 Lieberman-Aiden 实验室与 Juicer 和 Juicebox 这两款用于处理原位 Hi-C 数据的软件程序一同开发出来的。
随后,.pairs.gz、.cool、.mcool 等其他格式,由 4D Nucleome 数据协调与整合中心(4DN DCIC)开发并提出,目的是建立一个更灵活、更易读的可共享标准。这些格式已经在 NIH 资助的网络中被用于统一处理 Hi-C 数据,该网络的目标是理解基因组三维构象的动态变化。
在本文,我们会更详细地探讨这些最新的格式,以及在 Hi-C 数据处理过程中使用的其他现有标准和自定义格式。
数据预处理
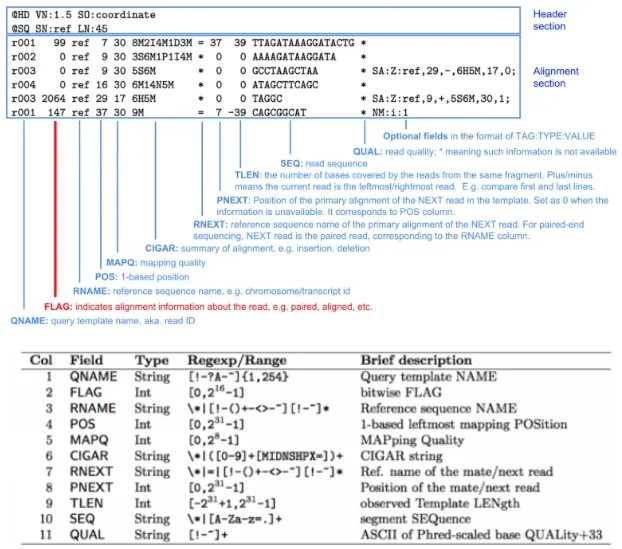
Hi-C 原始数据通常是用 FASTQ 格式生成的,这是一种广泛用于存储原始测序数据的标准格式。FASTQ 是一种文本格式,最早是在 Wellcome Trust Sanger 研究所开发出来的,用于存储原始测序读取以及它们的质量分数。原始序列会先与参考基因组序列进行比对,比对结果一般会存储在序列比对图(SAM)或二进制比对图(BAM)格式中,这两种格式最初是为 1000 基因组计划开发的。
SAM/BAM 文件中的比对结果会被进一步处理成一个接触列表文件,这个文件记录了所有代表有效相互作用位点的比对位置对。这种文件通常被称为“pairs”文件,比如 Juicer 生成的 merged_nodups.txt 文件,还有 4DN 使用的 PAIRS(.pairs.gz)文件。
接触列表文件中的条目会被聚合到基因组 bin 中,从而创建出一个接触矩阵文件。矩阵的 bin 或分辨率可以是千碱基对到兆碱基对长,也可能对应于限制性片段。像 .hic 和 .mcool 这样的最新格式能够携带多分辨率的矩阵,这样在使用交互式可视化工具(如 Juicebox、Juicebox.js 或 HiGlass)进行放大或缩小时,就可以快速切换分辨率了。.hic、.cool 和 .mcool 格式还包含了预计算的归一化向量以及原始矩阵,方便快速检索归一化矩阵。归一化可以控制由各种因素引起的偏差,比如限制性位点的不均匀分布、可比对性和 G+C 含量。最新的矩阵格式被设计成可以存储通过矩阵平衡计算出的乘法或除法向量,从而确保行和列的总和大致相等,而不是采用显式的偏差建模方法。
比对
SAM/BAM 是存储比对结果时常用的格式。SAM 是一种文本格式,BAM 则是 SAM 的压缩二进制版本。我们可以通过 Samtools 软件程序在 SAM 和 BAM 格式之间进行转换。像 BWA 和 Bowtie这样流行的短读比对器,能够生成 SAM 格式的输出,然后可以将其转换为 BAM 格式,从而实现更高效的存储。
在存储 Hi-C 数据的比对层面时,通常会考虑两个关键因素。第一是嵌合比对的记录,第二是两个末端的独立比对。
Hi-C 数据一般是配对末端的形式,两个末端分别代表两个相互作用的位点。在大多数情况下,连接点位于两个测序末端之间,此时两个末端会与参考基因组进行全长比对。不过,在大约 20 - 30% 的情况下,连接点会出现在两个测序末端中的一个内部,这就导致读段被分成两个不同的基因组位点。这些嵌合读段通常会在确认其有效性后被恢复,比如比对结果与单一真实的连接事件相符,尽管在定义有效性的标准上有所不同。通过这种方式,可以增加进入接触矩阵的有用读段数量。SAM/BAM 格式允许以剪切比对的形式来存储与嵌合读段比对相关的信息。
那些为通用测序数据开发的比对工具,通常会把一对读段看作是基因组中一段连续区域的两端。比如,BWA MEM在默认参数下,如果一对读段中的一个末端与参考基因组比对得很好,它可能会强行让另一个比对效果不好的末端也进行比对。然而,对于 Hi-C 数据来说,这种假设可能并不成立。为了避免这种情况,人们通常采用一种方法来比对 Hi-C 数据,即把两个末端分别当作独立的单末端数据来进行比对,然后再以某种方式把它们合并起来。不过,这种方法也有缺点,它往往会生成一个不完整或者不符合标准的 SAM/BAM 格式,与配对信息相关的字段和标志要么缺失,要么没有正确标注。4DN DCIC 提出了一个解决方案,即用参数 -SP5M 来运行 BWA MEM,从而创建一个 BAM 文件。这样,一对读段中的两个配对端在比对时会被独立对待,但输出的文件会被正确地格式化为一个配对末端的 BAM 文件。4DN Hi-C 数据处理流程就采用了这种方法。







