
引言
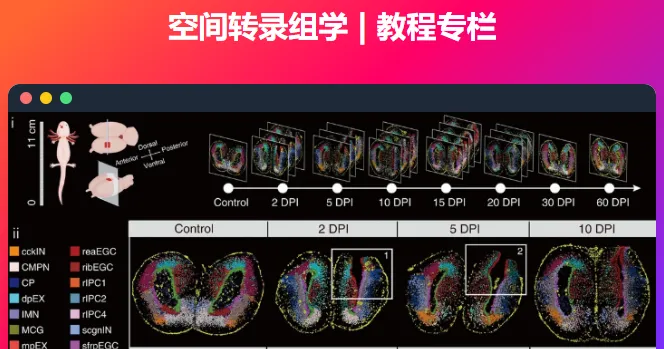
本系列讲解 空间转录组学 (Spatial Transcriptomics) 相关基础知识与数据分析教程,持续更新,欢迎关注,转发,文末有交流群!
质控
质量控制( QC )的目标是在进一步分析之前去除低质量spots或技术伪影。低质量spots可能由于文库制备或其他实验步骤中的问题而产生。为了防止将不必要的偏倚引入下游分析,低质量spot通常在进一步分析之前被移除。
低质量spots可以根据若干特征识别,包括:
- 文库大小(即每个spots的总独特分子标识符( UMI )计数)
- 表达特征的数量(即每个spots中非零 UMI 计数的基因数量)
- 比对到线粒体基因的读段比例(高比例指示细胞损伤)
- 每个spots的细胞数量(异常高的数值可能指示由于细胞分割这一计算步骤失败,或组织损伤导致的问题)
低文库大小或低表达特征数量可能表明 mRNA 捕获效率差,例如由于细胞损伤导致 mRNA 缺失,或反应效率低。高比例的线粒体读段可能指示细胞损伤,例如部分细胞裂解导致泄漏和胞质 mRNA 缺失,由此产生的读段因而集中在相对受线粒体膜保护的剩余线粒体 mRNA 上。异常高的每个spots细胞数量可能指示细胞分割步骤中的问题。
挑战
上述前三个特征是在 scRNA-seq 数据中使用的标准 QC 指标,它们通常也适用于 ST 数据。然而,在解释这些指标时必须谨慎,因为在基于测序的 ST 数据中,它们容易受到生物学的混淆。
例如,在大脑中,神经元胞体位于灰质,而白质几乎完全由神经元突起组成。这自然导致灰质区域检测到的基因数量和总体捕获的转录本数量远高于白质。此外,白质往往比灰质显示更高比例的线粒体读段。缺乏这一生物学背景,人们可能错误地认为白质区域质量低,应在下游分析前移除。
本文结构
在本系列中,我们将首先介绍使用各种策略识别低质量spots的方法,包括:
- 通过全局阈值法为单核 RNA-seq( snRNA-seq )开发的标准方法;
- 旨在缓解因空间混淆导致的 QC 偏倚的较新方法。
包 依赖
在本章中,我们将依赖以下包:
library(STexampleData)
library(ggspavis)
library(scater)
library(scuttle)
library(SpotSweeper)
library(patchwork)
数据导入
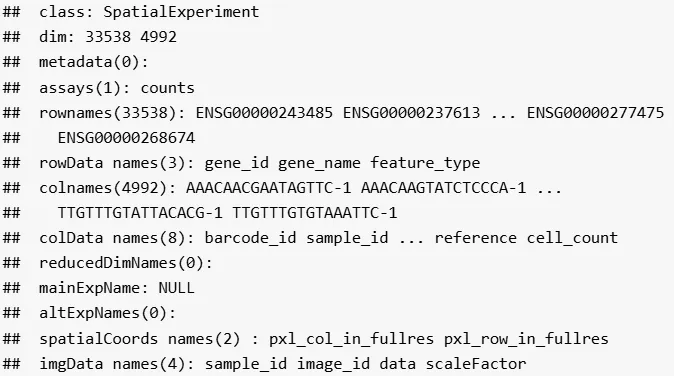
接下来,我们导入一个 10x Genomics Visium 数据集。该数据集先前已通过 R 之外工具的数据预处理流程进行了预处理,并以 SpatialExperiment 格式保存,可从 STexampleData 包下载。
该数据集包含来自一位供体的一个样本(Visium 捕获区域),由来自背外侧前额叶皮层(DLPFC)大脑区域的人类死后脑组织构成。
(spe <- Visium_humanDLPFC())
计算 QC 指标
我们结合 scater 包中的方法(适用于 scRNA-seq 数据的指标,这里把spots视作细胞)以及我们自写的函数,来计算上述 QC 指标。随后可用这些指标识别低质量spots。
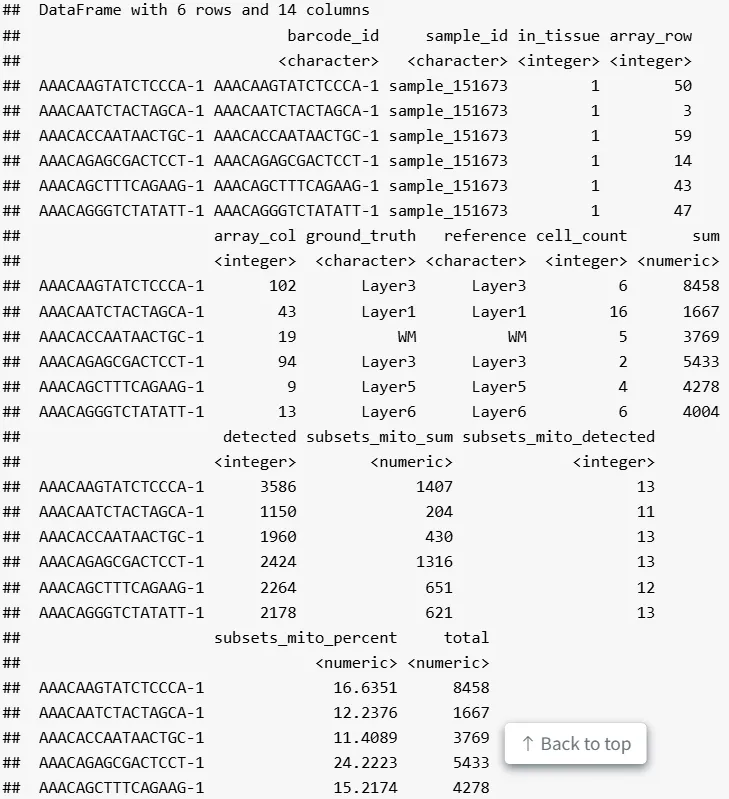
来自 scater 的 QC 指标可通过 addPerCellQC 函数计算,并添加到 SpatialExperiment 对象的列数据中。其中,sum 列包含每个spots的独特分子标识符( UMI )总数,detected 列包含每个spots检测到的独特基因数目,subsets_mito_percent 列则包含每个spots比对到线粒体基因的转录本百分比(或比例)。
首先,我们将对象做子集化,仅保留被组织切片覆盖的spots。其余spots为背景spots,我们不感兴趣,因为它们几乎全部由线粒体基因以及细胞碎片中的转录本构成。
# subset to keep only spots over tissue
spe <- spe[, spe$in_tissue == 1]
dim(spe)
## [1] 33538 3639
# identify mitochondrial genes
is_mito <- grepl("(^MT-)|(^mt-)", rowData(spe)$gene_name)
table(is_mito)
## is_mito
## FALSE TRUE
## 33525 13
rowData(spe)$gene_name[is_mito]
## [1] "MT-ND1" "MT-ND2" "MT-CO1" "MT-CO2" "MT-ATP8" "MT-ATP6" "MT-CO3"
## [8] "MT-ND3" "MT-ND4L" "MT-ND4" "MT-ND5" "MT-ND6" "MT-CYB"
# calculate per-spot QC metrics and store in colData
spe <- addPerCellQC(spe, subsets = list(mito = is_mito))
head(colData(spe))
未完待续,欢迎关注!





